GB 6227.1-1999
基本信息
标准号: GB 6227.1-1999
中文名称:食品添加剂 日落黄
标准类别:国家标准(GB)
英文名称:Food additive--Sunset yellow
标准状态:已作废
发布日期:1999-07-01
实施日期:2000-01-01
作废日期:2011-02-21
下载格式:pdf zip

标准分类号
标准ICS号: 食品技术>>香料和调料、食品添加剂>>67.220.20食品添加剂
中标分类号:食品>>食品添加剂与食用香料>>X42合成食品添加剂
出版信息
出版社:中国标准出版社
书号:155066.1-16252
页数:出版社:
标准价格:26.0
出版日期:2004-07-19
相关单位信息
首发日期:1986-03-26
复审日期:2004-10-14
起草单位:上海染料研究所
归口单位:全国食品添加剂标准化技术委员会
发布部门:国家质量技术监督局
主管部门:国家标准化管理委员会
标准简介
本标准规定了食品添加剂日落黄的要求、试验方法、检验规则以及标志、包装、运输、贮存。
标准图片预览




标准内容
GB 6227.1—1999
本标准非等效采用《日本食品添加物公定书》第6版(1992)。根据该书中“食用黄色5号(日落黄)”标推对GB6227.1-1995进行了修订。本标准同日本标准主要技术差异如下:1.本标准中含量指标为≥85.0%和含量≥60.0%的保留规格,日本含量指标≥85.0%。2.本标准日落黄85中干燥减量、氯化物(以NaCl计)及硫酸盐(以Na,SO。计)总量指标为≤15.0%,日本标推分列为干燥减量指标为≤10.0%,氯化物及硫酸盐指标为≤5.0%。3.本标准中重金属(以Pb计)含量指标为≤0.001%,日本指标为≤0.002%。4.本标推中副染料含量测定延用GB6227.1一1995,采用FAO/WHO中的测定方法,指标为≤4.0%。
5.本标准中碑含量测定采用GB/T8450—1987食品添加物中碑的测定方法,指标为≤0.0001%(以As计),日本指标为≤0.0004%(As2O.)。6。本标准中含量测定除三氯化钛滴定法外,增加相对简便的分光光度法,用于日常测定。以三氯化钛法作为仲裁方法。
7.本标准中氯化物(以NaCl计)及硫酸盐(以Na2SO。计)的测定方法为化学滴定法,日本标准采用离子色谱法。
本标准与GB6227.1-1995主要区别如下:1.GB6227.1--1995设日落黄85、日落黄60两个规格,本标准日落黄60为保留规格。2.本标准日落黄85规格中干燥减量、氯化物(以NaCl计)及硫酸盐(以Na2SO.计)合并为总量,指标为≤15.0%。
3.取消了异丙醚萃取物含量项目。其余与GB6227.1—1995相同。
本标准于1986年首次发布:于1999年进行第二次修订。本标准从实施之日起,同时代替GB6227.1一1995。本标准中附录A是标准的附录。
本标雄由国家石油和化学工业局提出。本标准由全国染料标准化技术委员会、卫生部食品监督检验所归口。本标准由上海市染料研究所、上海市卫生局卫生监督所负责起草。本标准主要起草人:丁德毅、刘静、施怀炯、成春虹、关建雄、周艳琴。本标准委托全国染料标准化技术委员会负责解释。416
1范围
中华人民共和国国家标准
食品添加剂
日落黄
Food additive-
Sunset yellow
GB6227.1—1999
代替GB6227.111995
本标谁规定了食品添加剂日落黄的要求、试验方法、检验规则以及标志、包装、运输、贮存。本标准适用于对氨基苯磺酸经重氮化后与2-酚-6-磺酸钠偶合而成的染料。结构式:
分子式:CH.N,O,S2Na2
相对分子质量:452.37(按1995年国际相对原子质量)2引用标准
下列标准所包含的条文,通过在本标推中引用而构成为本标推的条文。本标准出版时,所示版本均为有效。所有标准都会被修订,使用本标谁的各方应探讨使用下列标准最新版本的可能性。GB/T601—1988化学试剂滴定分析(容量分析)用标准溶液的制备化学试剂杂质测定用标准溶液的制备GB/T 602—1988
GB/T603—1988化学试剂试验方法中所用制剂及制品的制备GB/T6682—1992分析实验室用水规格和试验方法GB/T 8450—1987
3要求
食品添加剂中碑的测定方法
3.1外观橙红色粉末。
3.2食品添加剂日落黄85的质量应符合表1要求。国家质量技术监督局1999-07-12批准2000-01-01实施
GB6227.1 - 1999
表1要求
于燥减、氯化物(以NaCl计)及硫酸盐(以NazSO,计)总量水不溶物
副染料
砷(以As计)
重金属(以Pb计)
食品添加剂日落黄60的质量应符合表2要求,日落黄60为保留规格。3.3
表2要求
干燥减量
永不溶物
副染料
砷(以As计)
重金属(以Pb计)
注:保留规格为限制发展、准备取消的规格。4
试验方法
本标准所用试剂和水,在没有注明其他要求时,均指分析纯试剂和GB/T6682规定的三级水。试验中所需标准溶液、杂质标准溶液、制剂及制品在没有注明其他规定时,均按GB/T601、GB/T602、GB/T603之规定配制。
4.1外观
用目视测定
4.2鉴别
4.2.1试剂和材料
a)硫酸溶液:1+100;
b)乙酸铵溶液:1.5g/L,
4.2.2仪器、设备
分光光度计。
4.2.3试验方法
4.2.3.1称取约0.1g试样,溶于100mL水中,呈橙色澄清溶液4.2.3.2取4.2.3.1中橙色的溶液40mL,加入硫酸溶液10mL后该溶液呈橙红色,取此液2~3滴加人5ml水中,显橙黄色溶液。
4.2.3.3称取0.1g试样,溶于100mL乙酸铵溶液,取此溶液1mL加乙酸铵溶液配至100ml,该溶液的最大吸收波长为482nm士2nm。4.3含量的测定
4.3.7三氯化钛滴定法(仲裁法)4.3.1.1方法提要
GB 6227. 1 1999
在碱性介质中,试样中偶氮基被三氯化钛还原分解,按三氯化钛标准滴定溶液的消耗量,来计算染料的百分含量。
4.3.1.2试剂和材料
a)酒石酸氢钠;
b)三氯化钛标推滴定溶液c(TiCl)=0.1mol/L(新配制,配制方法见附录A);c)钢瓶装二氧化碳。
4.3.1.3测试方法
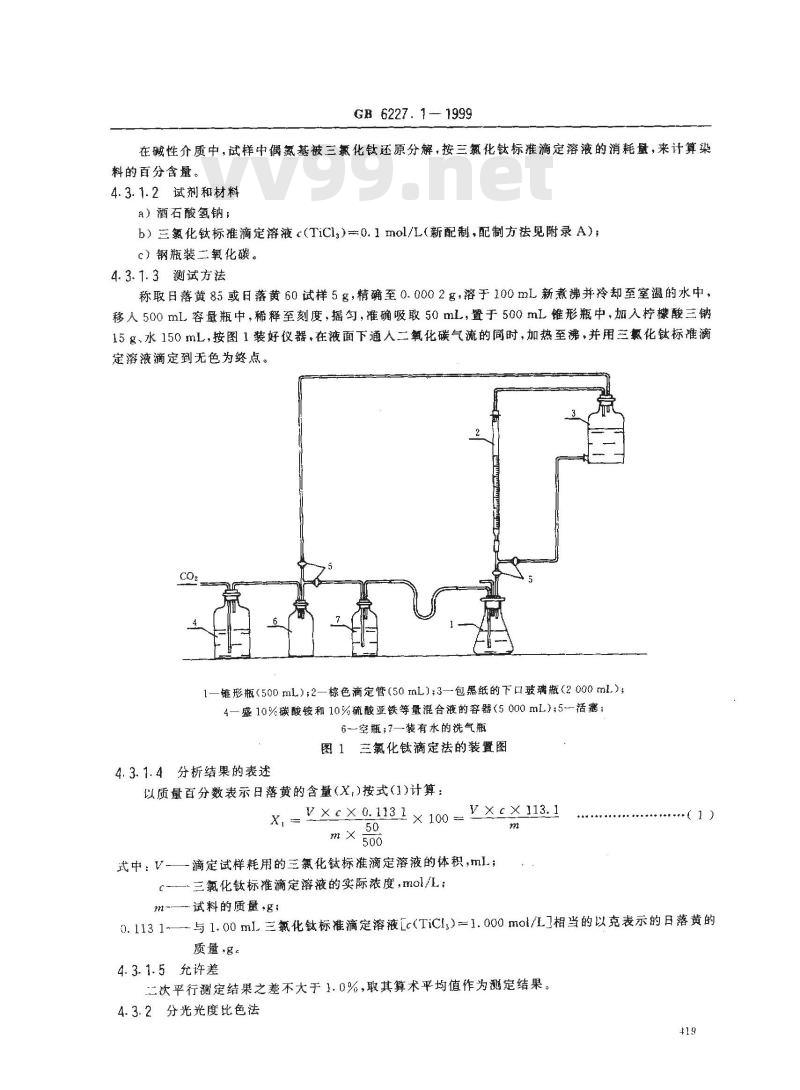
称取日落黄85或日落黄60试样5g,精确至0.0002g,溶于100mL新煮沸并冷却至室温的水中,移人500mL容量瓶中,稀释至刻度,摇匀,准确吸取50mL,置于500mL锥形瓶中,加入柠酸三钠15g、水150mL,按图1装好仪器,在液面下通人二氧化碳气流的同时,加热至沸,并用三氯化钛标准滴定溶液滴定到无色为终点。
锥形瓶(500ml);2一棕色滴定管(50mL);3一包黑纸的下口玻璃瓶(2000mL)4—盛10%碳酸铵和10%硫酸亚铁等量混合液的容器(5000mL);5-活塞:6一空瓶;7一装有水的洗气瓶
图1三氯化钛滴定法的装置图
4.3.1.4分析结果的表述
以质量百分数表示日落黄的含量(X,)按式(1)计算:Xx -XcX0.1131 × 100
V Xcx 113. 1
式中:V一滴定试样耗用的三氯化钛标准滴定溶液的体积,ml;c-—三氯化钛标准滴定溶液的实际浓度,mol/L;12
m-—试料的质量?g;
0.1131~—与1.00m三氯化钛标准滴定溶液L(TiCI,)=1.000mol/L1相当的以克表示的日落黄的质量,g。
4.3.1.5允许差
二次平行测定结果之差不大于1.0%,取其算术平均值作为测定结果。4.3.2分光光度比色法
4.3.2.1方法提要
GB 6227. 1-- 1999
将试样与已知含量的标样分别用水溶解后,在最大吸收波长处,分别测其吸光度,然后计算出试样的含量。
4.3.2.2试剂和材料
日落黄85或日落黄60标样:自制,含量以三氟化钛滴定法测定。4.3.2.3仪器、设备
a)分光光度计;
b)比色皿:10mm。
4.3.2.4日落黄85或日落黄60标样溶液的配制称取日落黄85或日落黄60标准样品0.5g,精确至0.0002g,溶于适量水中,移人1000mL容量瓶中,稀释至刻度,摇匀。准确吸取10mL,移人500mL容量瓶中,稀释至刻度,摇勾。4.3.2.5日落黄85或日落黄60试验溶液的配制称取试样0.5g,其余同标样溶液的配制。4.3.2.6测试方法
将标样溶液和试样溶液置于10mm比色血中,在482nm士2nm波长处用分光光度计测定各自的吸光度。
以水作参比液
4.3.2.7分析结果的表述
以质量百分数表示日落黄85或日落黄60的含量(X)按式(2)计算:X
式中:A—--试样溶液的吸光度;A.—标样溶液的吸光度;
X—日落黄85或日落黄60标准样品的质量百分含量(三氯化钛法)。4.3.2.8允许差
二次平行测定结果之差不大于2.0%,取其算术平均值作为测定结果。4.4干燥减量、氯化物(以NaCI计)及硫酸盐(以Na2SO,计)总量的测定。4.4.1干燥减量的测定
4.4.1.1测试方法
称取约2g试样,精确至0.01g,置于已恒重的(30~40)mm的称量瓶中,在135℃土2C恒温烘箱中烘至恒重。
4.4.1.2分析结果的表述
以质量百分数表示干燥减量的含量(X。)按式(3)计算:X
式中:m2——试料干燥前的质量·g,m
试料干燥至恒重后的质量,g。
m×100
4.4.7.3允许差
二次平行测定结果之差不大于0.2%,取其算术平均值作为测定结果。4.4.2氯化物(以NaCI计)含量的测定4.4.2.1试剂和材料
a)活性炭;
b)硝基苯;
c)硝酸溶液::1+1;
GB6227、1-1999
d)硝酸银标准滴定溶液:c(AgNO:)=0.1mol/L;e)硫氰酸铵标准滴定溶液:c(NH,CNS)=0.1mol/I;f)硫酸铁铵溶液;
配制:称硫酸铁铵14g,溶于水100mL,过滤,加硝酸10mL,贮于棕色瓶中。4.4.2.2试验溶液的配制
称取日落黄85试样约2g,精确至0.01g+加水200mL,加活性炭10g,再加1mL硝酸溶液,搅拌均匀,放置30min(其间不时搅动)用干燥滤纸过滤,如滤液有色,则加2g活性炭不时搅动下放置1h,再用干爆滤纸过滤,如仍有色则更换活性炭重新操作。4.4.2.3测试方法
移取以上试验溶液50mL,置于500mL锥形瓶中,加硝酸溶液2mL和硝酸银标准滴定溶液10ml.(氯化物含量多时要多加些)及硝基苯5mL,剧烈摇动到氯化银凝结,加入硫酸铁铵溶液1ml,用硫氰酸铵标准滴定溶液滴定至溶液呈砖红色邸为终点并保持1min。同时以同样方法做一空白试验。4.4.2.4分析结果的表述
以质量百分数表示氯化物(以NaCI计)含量(X)按式(4)计算:X, = -V)× 0. 058 4 × 100 = (V- V) ×c× 23. 36m×200
式中:V—滴定试样耗用硫氰酸铵标准滴定溶液的体积,mL;V:——-滴定空白溶液耗用硫氰酸铵滴定溶液的体积,mL:-硫氰酸铵标准滴定溶液的实际浓度,mol/I;-
m———试料质量,g;
0.0584——--与1.00mL硫氰酸铵标准滴定溶液[c(NH,CNS)1.000mal/L]相当的以克表示的氯化钠质量。
4.4.2.5允许差
二次平行测定结果之差不大于0.3%,取其算术平均值作为测定结果。4.4.3硫酸盐(以NVa2SO.计)含量的测定4.4.3.1试剂和材料
a)氨水;
b)氢氧化钠溶液:0.2g/L;
c)盐酸溶液:1+99:Vv99.net
d)乙醇:95%:
e)四羟基苯醒二钠-氯化钾:混合指示剂等量混合;f)硫酸标准滴定溶液:c(1/2H,SO4)=0.1mol/L;g)酚酰指示液:10g/L;
h)玫瑰红酸钠指示液:称取玫瑰红酸钠0.1g,溶于水10mL(现配现用);i)氯化钡标准滴定溶液:c(1/2BaCl2)=0.1mol/L;一配制:称取氯化钡12.25g,溶于水500mL,移人1000mL容量瓶中,稀释至刻度,摇勾。一标定:吸取硫酸标准溶液20mL,加水50mL,并用氨水中和到亮黄试纸呈碱性反应,然后用氯化钡标准溶液滴定,以玫瑰红酸钠指示液作液外指示,在滤纸上呈现玫瑰红色斑点保持2min不退为终点。
氯化钡标推滴定溶液的浓度(X:)按式(5)计算:XgKxc
.(5)
式中:V-
GB 6227. 1—1999
硫酸标准滴定溶液体积,mL:
氯化钡标准滴定溶液体积,mL;硫酸标准滴定溶液的实际浓度,mol /L。4.4.3.2测试方法
吸取试验溶液25mL,置于250mL锥形瓶中,加酚酸指示液1滴,滴加氢氧化钠溶液呈粉红色,然后滴加盐酸溶液到粉红色消失,再加乙醇30mL和四羟基苯醒二钠-氯化钾混合指示剂0.4g,摇勾,溶解后在不断摇动下以氮化钡标准溶液滴定到溶液呈玫瑰红色为终点,在滴定时要用灯光从侧面照射仔细观察,另用玫瑰红酸钠指示液作液外指示作比较,同时以相同方法做空白试验。4.4.3.3分析结果的表述
以质量百分数表示硫酸盐(以Na2SO,计)的含量(X。)按式(6)计算:(V -V) X c X 0. 071 x
(V - V) X cX 56. 8
式中:V-
.(6)
滴定试样溶液耗用氯化锁标准滴定溶液的体积,mL;滴定空白溶液耗用氯化钡标准滴定溶液的体积,mL;氯化钡标雄滴定溶液的实际浓度,mol/I试料的质量,g:
与1.00mL氯化钡标准滴定溶液[c(1/2BaCl.)=1.000mol/L]相当的以克表示的硫酸钠0. 071—
质量,g。
4.4.3.4允许差
二次平行测定结果之差不大于0.2%,取其算术平均值作为测定结果。·4.4.4分析结果的表述
以质量百分数表示干燥减量的含量、氯化物(以NaCI计)的含量及硫酸盐(以Na2SO,计)的含量的总和(X,))按式(7)计算:
X,= X +X +X.
式中:X,-
以质量百分数表示干燥减量的含量,%;X。——以质量百分数表示氯化物的含量,%;X,—以质量百分数表示硫酸盐的含量,%。4.5水不溶物含量的测定
4.5.1测定方法
称取约3g试样,精确至0.01g,置于500mL烧杯中,加人50℃~60C水250mL使之溶解,用已在135E士2C烘至恒重的4号玻璃砂芯过滤,并用热水充分洗涤到洗涤液无色,在135C士2℃恒温烘箱中烘至恒重。
4.5.2分析结果的表述
以质量百分数表示水不溶物的含量(X。)按式(8)计算:m2×100
式中:m3--干燥后水不溶物的质量·g试料的质量,g。
4.5.3允许差
二次平行测定结果之差不大于0.1%,取其算术平均值作为测定结果。4.6副染料含量的测定
4.6.1方法提要
GB 6227. 1— 1999
用纸上层析法将各组分分离、洗脱,然后用分光光度法定量。4.6.2试剂和材料
a)无水乙醇
b)正丁醇;
r)丙酮溶液:1+1;
d)氨水溶液:4+96;
e)碳酸氢钠溶液:4g/L。
4.6.3仪器、设备
a)分光光度计:
b)层析滤纸:1号中速,150mm×250mm;c)层析缸:$240mmX300mm;
d)微量进样器:100μL。
4.6.4测试方法
4.6.4.7纸上层析条件
a)展开剂:正丁醇+无水乙醇+氨水溶液=6+2+3;
b)温度:20C~25C。
4.6.4.2试样洗出液的配制
称取日落黄85或日落黄60试样1g,精确至0.01g,置于烧杯中、加人适量水溶解后,移人100ml.容量瓶中,稀释至刻度,摇句,用微量进样器吸取200μL,均匀地点在离滤纸底边25mm的一条基线上,成-直线,使其溶液在滤纸上的宽度不超过5mm,长度为130mm,用吹风机吹干,将滤纸放人层析缸中展开,滤纸底边浸入展开剂液面下10mm,待展开剂前沿线上升至150mm或直到副染料分离满意为止,取出层析滤纸,用吹风机以冷风吹干同时用空白滤纸在相同条件下展开(该空白滤纸必须和试验溶液展开用的滤纸在同一张600mmX600mm的滤纸上相邻部位裁取)。150mm
-副染料(1)
副染料(2)
主染料
一副染料(3)
副染料(4)
图2副染料层析示意图
GB 6227. 1—1999
将各个副染料和在空白滤纸上与各副染料相对应的部位的滤纸、按同样大小剪下,并剪成约5mmX15mm的细条,分别置于50mL的纳氏比色管中,各推确加人丙酮溶液5mL,摇动3min~5min后,再准确加入碳酸氢钠溶液20mI.充分摇动,将萃取液分别在3号玻璃砂芯漏斗中自然过滤,滤液必须澄清,无悬浮物,在各自副染料的最大吸收波长处,用50mm比色Ⅲ,在分光光度计上测定吸光度。以丙酮溶液5mI.和碳酸氢钠溶液20mL混合液作参比液。4.6.4.3标样洗出液的配制
准确吸取上述1%试样溶液2mL移入100mL容量瓶中,稀释至刻度,摇勾。用微量进样器吸取200μl,均匀地点在离滤纸底边25mm的一条基线上,用冷风吹干,将滤纸放入层析缸中展开,待展开剂前沿线仅上升40mm,取出后吹干,剪下所有染料部分,萃取操作同前,用厚度为10mm比色Ⅲl在最大吸收波长处测吸光度
同时用空白滤纸在相同条件下展开,按相同方法操作后测萃取液的吸光度。4.6.4.4分析结果的表述
以质量百分数表示副染料的含量(X。)按式(9)计算:(At - b)- :
..... -- (A. - bn)
式中: A.*,A.-
各副染料萃取液以50mm光径长度计算的吸光度;各副染料对照空白萃取液以50mm光径长度计算的吸光度;标准萃取液以10mm光径长度计算的吸光度;6.—标准对照空白萃取液以10mm光径长度计算的吸光度;5---折算成以10mm光径长度计算的比数;2-
一以1%试样溶液作基准的标准萃取液的参比浓度,%;S
4.6.4.5允许差
一试料的总含量。
二次平行测定结果之差不大于0.2%,取其算术平均值作为测定结果。4.7砷含量的测定
4.7.1试剂和材料
a)硝酸
b)硫酸溶液:1+l;
(9)
c)硝酸-高氯酸混合液:3+1;
d)砷标准溶液:0.001mg/mL。取含砷(As)0.1mg/mL.的标推溶液1mL于100mL容量瓶中,稀释至刻度。
4.7.2仪器、设备
按GB/T8450—1987中斑法的装置。4.7.3测试方法
称取日落黄 85或日落黄 60试样1.0g,精确至0.01g,置于圆底烧瓶中,加硝酸1.5mL和硫酸溶液5mI,用小火加热赶出二氧化氮气体,待溶液变成棕色,停止加热,放冷后加人硝酸-高氯酸混合液5mI.,强火加热直至溶液呈透明无色或微黄色,如仍不透明,放冷后再补加硝酸-高氯酸混合液5ml.,继续加热至溶液澄清无色或微黄色并产生白烟,停止加热,放冷后加水5mL加热至沸,除去残余的硝酸-高氯酸(必要时可再加水煮沸一次),继续加热至发生白烟,保持10min,放冷后移人100mL锥形瓶中,以下按GB/T8450—1987中2、4的规定进行。4.8重金属含量的测定
4.8.1试剂和材料
a)硫酸;
b)盐酸;
c)盐酸溶液:1+3;
d)乙酸溶液:1+3
e)氨水溶液:1十2;
1)硫化钠溶液:100g/L,
GB 6227. 1 -- 1999
g)铅标准溶液:0.01mg/mL。取含铅(Pb)0.1mg/mL的铅标准溶液10mL于100mL容量瓶中,稀释至刻度
4.8.2试样液的配制
称取日落黄85或日落黄60试样2.5g,精确至0.01g,置于用白金制(或石英制、瓷制)的中,加少量硫酸润湿,缓慢灼烧,尽量在低温下使之几乎全部灰化,再加硫酸1mL,逐渐加热至硫酸蒸气差不多不再发生。放人电炉中,在450℃~550℃灼烧至灰化,然后放冷。加盐酸3mL播勾,再加水7mL摇勾,用定量分析滤纸(5号C)过滤。用盐酸溶液5mL及水5mL洗涤滤纸上的残留物,将洗和滤液合并,加水配至50mL,作为试样液。用同样方法不加试样配制为空白试验液。4.8.3试验液的配制
量取试样液20mL,放人纳氏比色管中,加酚酸试液1滴,滴加氨水溶液至溶液显红色,再加乙酸溶液2ml,必要时过滤,用水洗滤纸,加水配至50mL,作为试验液。4.8.4比较液的配制
量取空白试验液20ml,放入纳氏比色管中,加入铅标准溶液2.0mL及酚指示液1滴,制备方法与试验液相同,作为比较液。4.8.5测试方法
在试验液和比较液中分别加入硫化钠溶液2滴,摇匀,放置5min后,试验液的颜色不得深于比较液。
5检验规则
5.1食品添加剂日落黄85或日落黄60,由生产单位的质量检验部门按本标准的规定对产品质量进行检验。生产单位应保证所有出厂的食品添加剂日落黄85或日落黄60质量均符合本标准的要求,并有定格式的质量证明书。
5.2食品添加剂日落黄85或日落黄60以一次拼混的均勾产品为一批。5.3瓶装产品采样应从每批产品包装箱(每箱为10×0.5kg)总数中选取10%,再从选出的箱中选取10%瓶,从选出的瓶中,在每瓶的中心处取出不少于50g的样品,取样时应小心,不使外界杂质落人产品中,将所采得样品迅速混匀后从中取约100g,分别装于两个清洁,干燥的磨口玻璃瓶中,并用石蜡密封,贴上标签,注明生产厂名、产品名称,批号、采样日期。一瓶供检验,另一瓶留样备查。5.4检验结果若有一项指标不符合本标准要求时,应重新自两倍量的包装中取样复检,复检结果如仍有一项指标不符合本标准要求时,则该批产品为不合格。5.5使用单位有权按照本标准规定的检验规则和试验方法对所收到的食品添加剂日落黄85或日落黄60的质量进行验收。
6标志、包装、运输、贮存
6.1包装箱上应有牢固清晰的标志,内容包括:“食品添加剂着色剂”字样、产品名称、商标、生产厂名和地址、数量、毛重、生产日期和批号、保质期。6.2每一瓶出厂产品,应有明显的标识,内容包括:“食品添加剂”字样、产品名称、生产厂名和地址、商425
GB6227.1—1999
标、生产和食品卫生许可证号、产品标准号和标准名称、保质期、生产日期和批号、净含量、使用说明。6.3食品添加剂日落黄85或日落黄60装于聚乙烯塑料瓶中,每瓶0.5kg,每10瓶外套箱固封,其他形式包装可由生产厂和用户协商确定。6.4运输时必须防雨、防潮、防晒,应贮存于干燥、阴凉的库房中。6.5本产品在运中不得与有毒、有害的其他物质混装、混运、一起堆放。6.6本产品从生产日期起,保质期为五年。逾期重新检验完全符合本标准的要求,方可使用。126
A1试剂和材料
a)盐酸:
b)硫酸亚铁铵;
c)硫氰酸铵溶液:200g/I;
d)硫酸溶液:1+1;
GB 6227.1—1999
附录A
(标准的附录)
三氯化钛标准滴定溶液的制备方法e)重铬酸钾标准滴定溶液:c(1/6K.Cr.0,)=0.1mol/L。A2仪器、设备
见图1。
A3三氯化钛标准滴定溶液的制备A3.1配制
取三氯化钛溶液100mL和盐酸75mL置于1000mL棕色容量瓶中,用新煮沸并已冷却到室温的水稀释至刻度,摇勾,立即倒人避光的下口瓶中,在二氧化碳气体保护下贮藏。A3.2标定
称取硫酸亚铁铵3g,精确至0.0002g,置于500mL锥形瓶中,在二氧化碳气流保护作用下,加入新煮沸并已冷却的水50mL,使其溶解,再加入硫酸溶液25mI,继续在液面下通人二氧化碳气流作保护,迅速准确加入重铬酸钾标准滴定溶液45mI,然后用需标定的三氮化钛标准滴定溶液滴定到接近计算量终点,立即加人硫氰酸铵溶液25ml,并继续用需标定的三氯化钛标准滴定溶液滴定到红色转变为绿色,即为终点。整个滴定过程应在二氧化碳气流保护下操作,同时以45mI水代替重铬酸钾游液以相同的方法做一空白试验。
A3.3三氯化钛标准滴定溶液浓度的表述三氯化钛标准滴定溶液的浓度(c)按式(A1)计算:V,xci
(Al)
式中:V,一一重铬酸钾标准滴定溶液体积,mL;一滴定被重铬酸钾标准滴定溶液氧化成高铁所耗用的三氯化钛标准滴定溶液的体积,mI;Va
滴定空白耗用三氯化钛标推滴定溶液的体积,mL:Va
重铬酸钾标准滴定溶液的实际浓度,mal/L。注:以上标定需在分析样品时即时标定。427
小提示:此标准内容仅展示完整标准里的部分截取内容,若需要完整标准请到上方自行免费下载完整标准文档。
本标准非等效采用《日本食品添加物公定书》第6版(1992)。根据该书中“食用黄色5号(日落黄)”标推对GB6227.1-1995进行了修订。本标准同日本标准主要技术差异如下:1.本标准中含量指标为≥85.0%和含量≥60.0%的保留规格,日本含量指标≥85.0%。2.本标准日落黄85中干燥减量、氯化物(以NaCl计)及硫酸盐(以Na,SO。计)总量指标为≤15.0%,日本标推分列为干燥减量指标为≤10.0%,氯化物及硫酸盐指标为≤5.0%。3.本标准中重金属(以Pb计)含量指标为≤0.001%,日本指标为≤0.002%。4.本标推中副染料含量测定延用GB6227.1一1995,采用FAO/WHO中的测定方法,指标为≤4.0%。
5.本标准中碑含量测定采用GB/T8450—1987食品添加物中碑的测定方法,指标为≤0.0001%(以As计),日本指标为≤0.0004%(As2O.)。6。本标准中含量测定除三氯化钛滴定法外,增加相对简便的分光光度法,用于日常测定。以三氯化钛法作为仲裁方法。
7.本标准中氯化物(以NaCl计)及硫酸盐(以Na2SO。计)的测定方法为化学滴定法,日本标准采用离子色谱法。
本标准与GB6227.1-1995主要区别如下:1.GB6227.1--1995设日落黄85、日落黄60两个规格,本标准日落黄60为保留规格。2.本标准日落黄85规格中干燥减量、氯化物(以NaCl计)及硫酸盐(以Na2SO.计)合并为总量,指标为≤15.0%。
3.取消了异丙醚萃取物含量项目。其余与GB6227.1—1995相同。
本标准于1986年首次发布:于1999年进行第二次修订。本标准从实施之日起,同时代替GB6227.1一1995。本标准中附录A是标准的附录。
本标雄由国家石油和化学工业局提出。本标准由全国染料标准化技术委员会、卫生部食品监督检验所归口。本标准由上海市染料研究所、上海市卫生局卫生监督所负责起草。本标准主要起草人:丁德毅、刘静、施怀炯、成春虹、关建雄、周艳琴。本标准委托全国染料标准化技术委员会负责解释。416
1范围
中华人民共和国国家标准
食品添加剂
日落黄
Food additive-
Sunset yellow
GB6227.1—1999
代替GB6227.111995
本标谁规定了食品添加剂日落黄的要求、试验方法、检验规则以及标志、包装、运输、贮存。本标准适用于对氨基苯磺酸经重氮化后与2-酚-6-磺酸钠偶合而成的染料。结构式:
分子式:CH.N,O,S2Na2
相对分子质量:452.37(按1995年国际相对原子质量)2引用标准
下列标准所包含的条文,通过在本标推中引用而构成为本标推的条文。本标准出版时,所示版本均为有效。所有标准都会被修订,使用本标谁的各方应探讨使用下列标准最新版本的可能性。GB/T601—1988化学试剂滴定分析(容量分析)用标准溶液的制备化学试剂杂质测定用标准溶液的制备GB/T 602—1988
GB/T603—1988化学试剂试验方法中所用制剂及制品的制备GB/T6682—1992分析实验室用水规格和试验方法GB/T 8450—1987
3要求
食品添加剂中碑的测定方法
3.1外观橙红色粉末。
3.2食品添加剂日落黄85的质量应符合表1要求。国家质量技术监督局1999-07-12批准2000-01-01实施
GB6227.1 - 1999
表1要求
于燥减、氯化物(以NaCl计)及硫酸盐(以NazSO,计)总量水不溶物
副染料
砷(以As计)
重金属(以Pb计)
食品添加剂日落黄60的质量应符合表2要求,日落黄60为保留规格。3.3
表2要求
干燥减量
永不溶物
副染料
砷(以As计)
重金属(以Pb计)
注:保留规格为限制发展、准备取消的规格。4
试验方法
本标准所用试剂和水,在没有注明其他要求时,均指分析纯试剂和GB/T6682规定的三级水。试验中所需标准溶液、杂质标准溶液、制剂及制品在没有注明其他规定时,均按GB/T601、GB/T602、GB/T603之规定配制。
4.1外观
用目视测定
4.2鉴别
4.2.1试剂和材料
a)硫酸溶液:1+100;
b)乙酸铵溶液:1.5g/L,
4.2.2仪器、设备
分光光度计。
4.2.3试验方法
4.2.3.1称取约0.1g试样,溶于100mL水中,呈橙色澄清溶液4.2.3.2取4.2.3.1中橙色的溶液40mL,加入硫酸溶液10mL后该溶液呈橙红色,取此液2~3滴加人5ml水中,显橙黄色溶液。
4.2.3.3称取0.1g试样,溶于100mL乙酸铵溶液,取此溶液1mL加乙酸铵溶液配至100ml,该溶液的最大吸收波长为482nm士2nm。4.3含量的测定
4.3.7三氯化钛滴定法(仲裁法)4.3.1.1方法提要
GB 6227. 1 1999
在碱性介质中,试样中偶氮基被三氯化钛还原分解,按三氯化钛标准滴定溶液的消耗量,来计算染料的百分含量。
4.3.1.2试剂和材料
a)酒石酸氢钠;
b)三氯化钛标推滴定溶液c(TiCl)=0.1mol/L(新配制,配制方法见附录A);c)钢瓶装二氧化碳。
4.3.1.3测试方法
称取日落黄85或日落黄60试样5g,精确至0.0002g,溶于100mL新煮沸并冷却至室温的水中,移人500mL容量瓶中,稀释至刻度,摇匀,准确吸取50mL,置于500mL锥形瓶中,加入柠酸三钠15g、水150mL,按图1装好仪器,在液面下通人二氧化碳气流的同时,加热至沸,并用三氯化钛标准滴定溶液滴定到无色为终点。
锥形瓶(500ml);2一棕色滴定管(50mL);3一包黑纸的下口玻璃瓶(2000mL)4—盛10%碳酸铵和10%硫酸亚铁等量混合液的容器(5000mL);5-活塞:6一空瓶;7一装有水的洗气瓶
图1三氯化钛滴定法的装置图
4.3.1.4分析结果的表述
以质量百分数表示日落黄的含量(X,)按式(1)计算:Xx -XcX0.1131 × 100
V Xcx 113. 1
式中:V一滴定试样耗用的三氯化钛标准滴定溶液的体积,ml;c-—三氯化钛标准滴定溶液的实际浓度,mol/L;12
m-—试料的质量?g;
0.1131~—与1.00m三氯化钛标准滴定溶液L(TiCI,)=1.000mol/L1相当的以克表示的日落黄的质量,g。
4.3.1.5允许差
二次平行测定结果之差不大于1.0%,取其算术平均值作为测定结果。4.3.2分光光度比色法
4.3.2.1方法提要
GB 6227. 1-- 1999
将试样与已知含量的标样分别用水溶解后,在最大吸收波长处,分别测其吸光度,然后计算出试样的含量。
4.3.2.2试剂和材料
日落黄85或日落黄60标样:自制,含量以三氟化钛滴定法测定。4.3.2.3仪器、设备
a)分光光度计;
b)比色皿:10mm。
4.3.2.4日落黄85或日落黄60标样溶液的配制称取日落黄85或日落黄60标准样品0.5g,精确至0.0002g,溶于适量水中,移人1000mL容量瓶中,稀释至刻度,摇匀。准确吸取10mL,移人500mL容量瓶中,稀释至刻度,摇勾。4.3.2.5日落黄85或日落黄60试验溶液的配制称取试样0.5g,其余同标样溶液的配制。4.3.2.6测试方法
将标样溶液和试样溶液置于10mm比色血中,在482nm士2nm波长处用分光光度计测定各自的吸光度。
以水作参比液
4.3.2.7分析结果的表述
以质量百分数表示日落黄85或日落黄60的含量(X)按式(2)计算:X
式中:A—--试样溶液的吸光度;A.—标样溶液的吸光度;
X—日落黄85或日落黄60标准样品的质量百分含量(三氯化钛法)。4.3.2.8允许差
二次平行测定结果之差不大于2.0%,取其算术平均值作为测定结果。4.4干燥减量、氯化物(以NaCI计)及硫酸盐(以Na2SO,计)总量的测定。4.4.1干燥减量的测定
4.4.1.1测试方法
称取约2g试样,精确至0.01g,置于已恒重的(30~40)mm的称量瓶中,在135℃土2C恒温烘箱中烘至恒重。
4.4.1.2分析结果的表述
以质量百分数表示干燥减量的含量(X。)按式(3)计算:X
式中:m2——试料干燥前的质量·g,m
试料干燥至恒重后的质量,g。
m×100
4.4.7.3允许差
二次平行测定结果之差不大于0.2%,取其算术平均值作为测定结果。4.4.2氯化物(以NaCI计)含量的测定4.4.2.1试剂和材料
a)活性炭;
b)硝基苯;
c)硝酸溶液::1+1;
GB6227、1-1999
d)硝酸银标准滴定溶液:c(AgNO:)=0.1mol/L;e)硫氰酸铵标准滴定溶液:c(NH,CNS)=0.1mol/I;f)硫酸铁铵溶液;
配制:称硫酸铁铵14g,溶于水100mL,过滤,加硝酸10mL,贮于棕色瓶中。4.4.2.2试验溶液的配制
称取日落黄85试样约2g,精确至0.01g+加水200mL,加活性炭10g,再加1mL硝酸溶液,搅拌均匀,放置30min(其间不时搅动)用干燥滤纸过滤,如滤液有色,则加2g活性炭不时搅动下放置1h,再用干爆滤纸过滤,如仍有色则更换活性炭重新操作。4.4.2.3测试方法
移取以上试验溶液50mL,置于500mL锥形瓶中,加硝酸溶液2mL和硝酸银标准滴定溶液10ml.(氯化物含量多时要多加些)及硝基苯5mL,剧烈摇动到氯化银凝结,加入硫酸铁铵溶液1ml,用硫氰酸铵标准滴定溶液滴定至溶液呈砖红色邸为终点并保持1min。同时以同样方法做一空白试验。4.4.2.4分析结果的表述
以质量百分数表示氯化物(以NaCI计)含量(X)按式(4)计算:X, = -V)× 0. 058 4 × 100 = (V- V) ×c× 23. 36m×200
式中:V—滴定试样耗用硫氰酸铵标准滴定溶液的体积,mL;V:——-滴定空白溶液耗用硫氰酸铵滴定溶液的体积,mL:-硫氰酸铵标准滴定溶液的实际浓度,mol/I;-
m———试料质量,g;
0.0584——--与1.00mL硫氰酸铵标准滴定溶液[c(NH,CNS)1.000mal/L]相当的以克表示的氯化钠质量。
4.4.2.5允许差
二次平行测定结果之差不大于0.3%,取其算术平均值作为测定结果。4.4.3硫酸盐(以NVa2SO.计)含量的测定4.4.3.1试剂和材料
a)氨水;
b)氢氧化钠溶液:0.2g/L;
c)盐酸溶液:1+99:Vv99.net
d)乙醇:95%:
e)四羟基苯醒二钠-氯化钾:混合指示剂等量混合;f)硫酸标准滴定溶液:c(1/2H,SO4)=0.1mol/L;g)酚酰指示液:10g/L;
h)玫瑰红酸钠指示液:称取玫瑰红酸钠0.1g,溶于水10mL(现配现用);i)氯化钡标准滴定溶液:c(1/2BaCl2)=0.1mol/L;一配制:称取氯化钡12.25g,溶于水500mL,移人1000mL容量瓶中,稀释至刻度,摇勾。一标定:吸取硫酸标准溶液20mL,加水50mL,并用氨水中和到亮黄试纸呈碱性反应,然后用氯化钡标准溶液滴定,以玫瑰红酸钠指示液作液外指示,在滤纸上呈现玫瑰红色斑点保持2min不退为终点。
氯化钡标推滴定溶液的浓度(X:)按式(5)计算:XgKxc
.(5)
式中:V-
GB 6227. 1—1999
硫酸标准滴定溶液体积,mL:
氯化钡标准滴定溶液体积,mL;硫酸标准滴定溶液的实际浓度,mol /L。4.4.3.2测试方法
吸取试验溶液25mL,置于250mL锥形瓶中,加酚酸指示液1滴,滴加氢氧化钠溶液呈粉红色,然后滴加盐酸溶液到粉红色消失,再加乙醇30mL和四羟基苯醒二钠-氯化钾混合指示剂0.4g,摇勾,溶解后在不断摇动下以氮化钡标准溶液滴定到溶液呈玫瑰红色为终点,在滴定时要用灯光从侧面照射仔细观察,另用玫瑰红酸钠指示液作液外指示作比较,同时以相同方法做空白试验。4.4.3.3分析结果的表述
以质量百分数表示硫酸盐(以Na2SO,计)的含量(X。)按式(6)计算:(V -V) X c X 0. 071 x
(V - V) X cX 56. 8
式中:V-
.(6)
滴定试样溶液耗用氯化锁标准滴定溶液的体积,mL;滴定空白溶液耗用氯化钡标准滴定溶液的体积,mL;氯化钡标雄滴定溶液的实际浓度,mol/I试料的质量,g:
与1.00mL氯化钡标准滴定溶液[c(1/2BaCl.)=1.000mol/L]相当的以克表示的硫酸钠0. 071—
质量,g。
4.4.3.4允许差
二次平行测定结果之差不大于0.2%,取其算术平均值作为测定结果。·4.4.4分析结果的表述
以质量百分数表示干燥减量的含量、氯化物(以NaCI计)的含量及硫酸盐(以Na2SO,计)的含量的总和(X,))按式(7)计算:
X,= X +X +X.
式中:X,-
以质量百分数表示干燥减量的含量,%;X。——以质量百分数表示氯化物的含量,%;X,—以质量百分数表示硫酸盐的含量,%。4.5水不溶物含量的测定
4.5.1测定方法
称取约3g试样,精确至0.01g,置于500mL烧杯中,加人50℃~60C水250mL使之溶解,用已在135E士2C烘至恒重的4号玻璃砂芯过滤,并用热水充分洗涤到洗涤液无色,在135C士2℃恒温烘箱中烘至恒重。
4.5.2分析结果的表述
以质量百分数表示水不溶物的含量(X。)按式(8)计算:m2×100
式中:m3--干燥后水不溶物的质量·g试料的质量,g。
4.5.3允许差
二次平行测定结果之差不大于0.1%,取其算术平均值作为测定结果。4.6副染料含量的测定
4.6.1方法提要
GB 6227. 1— 1999
用纸上层析法将各组分分离、洗脱,然后用分光光度法定量。4.6.2试剂和材料
a)无水乙醇
b)正丁醇;
r)丙酮溶液:1+1;
d)氨水溶液:4+96;
e)碳酸氢钠溶液:4g/L。
4.6.3仪器、设备
a)分光光度计:
b)层析滤纸:1号中速,150mm×250mm;c)层析缸:$240mmX300mm;
d)微量进样器:100μL。
4.6.4测试方法
4.6.4.7纸上层析条件
a)展开剂:正丁醇+无水乙醇+氨水溶液=6+2+3;
b)温度:20C~25C。
4.6.4.2试样洗出液的配制
称取日落黄85或日落黄60试样1g,精确至0.01g,置于烧杯中、加人适量水溶解后,移人100ml.容量瓶中,稀释至刻度,摇句,用微量进样器吸取200μL,均匀地点在离滤纸底边25mm的一条基线上,成-直线,使其溶液在滤纸上的宽度不超过5mm,长度为130mm,用吹风机吹干,将滤纸放人层析缸中展开,滤纸底边浸入展开剂液面下10mm,待展开剂前沿线上升至150mm或直到副染料分离满意为止,取出层析滤纸,用吹风机以冷风吹干同时用空白滤纸在相同条件下展开(该空白滤纸必须和试验溶液展开用的滤纸在同一张600mmX600mm的滤纸上相邻部位裁取)。150mm
-副染料(1)
副染料(2)
主染料
一副染料(3)
副染料(4)
图2副染料层析示意图
GB 6227. 1—1999
将各个副染料和在空白滤纸上与各副染料相对应的部位的滤纸、按同样大小剪下,并剪成约5mmX15mm的细条,分别置于50mL的纳氏比色管中,各推确加人丙酮溶液5mL,摇动3min~5min后,再准确加入碳酸氢钠溶液20mI.充分摇动,将萃取液分别在3号玻璃砂芯漏斗中自然过滤,滤液必须澄清,无悬浮物,在各自副染料的最大吸收波长处,用50mm比色Ⅲ,在分光光度计上测定吸光度。以丙酮溶液5mI.和碳酸氢钠溶液20mL混合液作参比液。4.6.4.3标样洗出液的配制
准确吸取上述1%试样溶液2mL移入100mL容量瓶中,稀释至刻度,摇勾。用微量进样器吸取200μl,均匀地点在离滤纸底边25mm的一条基线上,用冷风吹干,将滤纸放入层析缸中展开,待展开剂前沿线仅上升40mm,取出后吹干,剪下所有染料部分,萃取操作同前,用厚度为10mm比色Ⅲl在最大吸收波长处测吸光度
同时用空白滤纸在相同条件下展开,按相同方法操作后测萃取液的吸光度。4.6.4.4分析结果的表述
以质量百分数表示副染料的含量(X。)按式(9)计算:(At - b)- :
..... -- (A. - bn)
式中: A.*,A.-
各副染料萃取液以50mm光径长度计算的吸光度;各副染料对照空白萃取液以50mm光径长度计算的吸光度;标准萃取液以10mm光径长度计算的吸光度;6.—标准对照空白萃取液以10mm光径长度计算的吸光度;5---折算成以10mm光径长度计算的比数;2-
一以1%试样溶液作基准的标准萃取液的参比浓度,%;S
4.6.4.5允许差
一试料的总含量。
二次平行测定结果之差不大于0.2%,取其算术平均值作为测定结果。4.7砷含量的测定
4.7.1试剂和材料
a)硝酸
b)硫酸溶液:1+l;
(9)
c)硝酸-高氯酸混合液:3+1;
d)砷标准溶液:0.001mg/mL。取含砷(As)0.1mg/mL.的标推溶液1mL于100mL容量瓶中,稀释至刻度。
4.7.2仪器、设备
按GB/T8450—1987中斑法的装置。4.7.3测试方法
称取日落黄 85或日落黄 60试样1.0g,精确至0.01g,置于圆底烧瓶中,加硝酸1.5mL和硫酸溶液5mI,用小火加热赶出二氧化氮气体,待溶液变成棕色,停止加热,放冷后加人硝酸-高氯酸混合液5mI.,强火加热直至溶液呈透明无色或微黄色,如仍不透明,放冷后再补加硝酸-高氯酸混合液5ml.,继续加热至溶液澄清无色或微黄色并产生白烟,停止加热,放冷后加水5mL加热至沸,除去残余的硝酸-高氯酸(必要时可再加水煮沸一次),继续加热至发生白烟,保持10min,放冷后移人100mL锥形瓶中,以下按GB/T8450—1987中2、4的规定进行。4.8重金属含量的测定
4.8.1试剂和材料
a)硫酸;
b)盐酸;
c)盐酸溶液:1+3;
d)乙酸溶液:1+3
e)氨水溶液:1十2;
1)硫化钠溶液:100g/L,
GB 6227. 1 -- 1999
g)铅标准溶液:0.01mg/mL。取含铅(Pb)0.1mg/mL的铅标准溶液10mL于100mL容量瓶中,稀释至刻度
4.8.2试样液的配制
称取日落黄85或日落黄60试样2.5g,精确至0.01g,置于用白金制(或石英制、瓷制)的中,加少量硫酸润湿,缓慢灼烧,尽量在低温下使之几乎全部灰化,再加硫酸1mL,逐渐加热至硫酸蒸气差不多不再发生。放人电炉中,在450℃~550℃灼烧至灰化,然后放冷。加盐酸3mL播勾,再加水7mL摇勾,用定量分析滤纸(5号C)过滤。用盐酸溶液5mL及水5mL洗涤滤纸上的残留物,将洗和滤液合并,加水配至50mL,作为试样液。用同样方法不加试样配制为空白试验液。4.8.3试验液的配制
量取试样液20mL,放人纳氏比色管中,加酚酸试液1滴,滴加氨水溶液至溶液显红色,再加乙酸溶液2ml,必要时过滤,用水洗滤纸,加水配至50mL,作为试验液。4.8.4比较液的配制
量取空白试验液20ml,放入纳氏比色管中,加入铅标准溶液2.0mL及酚指示液1滴,制备方法与试验液相同,作为比较液。4.8.5测试方法
在试验液和比较液中分别加入硫化钠溶液2滴,摇匀,放置5min后,试验液的颜色不得深于比较液。
5检验规则
5.1食品添加剂日落黄85或日落黄60,由生产单位的质量检验部门按本标准的规定对产品质量进行检验。生产单位应保证所有出厂的食品添加剂日落黄85或日落黄60质量均符合本标准的要求,并有定格式的质量证明书。
5.2食品添加剂日落黄85或日落黄60以一次拼混的均勾产品为一批。5.3瓶装产品采样应从每批产品包装箱(每箱为10×0.5kg)总数中选取10%,再从选出的箱中选取10%瓶,从选出的瓶中,在每瓶的中心处取出不少于50g的样品,取样时应小心,不使外界杂质落人产品中,将所采得样品迅速混匀后从中取约100g,分别装于两个清洁,干燥的磨口玻璃瓶中,并用石蜡密封,贴上标签,注明生产厂名、产品名称,批号、采样日期。一瓶供检验,另一瓶留样备查。5.4检验结果若有一项指标不符合本标准要求时,应重新自两倍量的包装中取样复检,复检结果如仍有一项指标不符合本标准要求时,则该批产品为不合格。5.5使用单位有权按照本标准规定的检验规则和试验方法对所收到的食品添加剂日落黄85或日落黄60的质量进行验收。
6标志、包装、运输、贮存
6.1包装箱上应有牢固清晰的标志,内容包括:“食品添加剂着色剂”字样、产品名称、商标、生产厂名和地址、数量、毛重、生产日期和批号、保质期。6.2每一瓶出厂产品,应有明显的标识,内容包括:“食品添加剂”字样、产品名称、生产厂名和地址、商425
GB6227.1—1999
标、生产和食品卫生许可证号、产品标准号和标准名称、保质期、生产日期和批号、净含量、使用说明。6.3食品添加剂日落黄85或日落黄60装于聚乙烯塑料瓶中,每瓶0.5kg,每10瓶外套箱固封,其他形式包装可由生产厂和用户协商确定。6.4运输时必须防雨、防潮、防晒,应贮存于干燥、阴凉的库房中。6.5本产品在运中不得与有毒、有害的其他物质混装、混运、一起堆放。6.6本产品从生产日期起,保质期为五年。逾期重新检验完全符合本标准的要求,方可使用。126
A1试剂和材料
a)盐酸:
b)硫酸亚铁铵;
c)硫氰酸铵溶液:200g/I;
d)硫酸溶液:1+1;
GB 6227.1—1999
附录A
(标准的附录)
三氯化钛标准滴定溶液的制备方法e)重铬酸钾标准滴定溶液:c(1/6K.Cr.0,)=0.1mol/L。A2仪器、设备
见图1。
A3三氯化钛标准滴定溶液的制备A3.1配制
取三氯化钛溶液100mL和盐酸75mL置于1000mL棕色容量瓶中,用新煮沸并已冷却到室温的水稀释至刻度,摇勾,立即倒人避光的下口瓶中,在二氧化碳气体保护下贮藏。A3.2标定
称取硫酸亚铁铵3g,精确至0.0002g,置于500mL锥形瓶中,在二氧化碳气流保护作用下,加入新煮沸并已冷却的水50mL,使其溶解,再加入硫酸溶液25mI,继续在液面下通人二氧化碳气流作保护,迅速准确加入重铬酸钾标准滴定溶液45mI,然后用需标定的三氮化钛标准滴定溶液滴定到接近计算量终点,立即加人硫氰酸铵溶液25ml,并继续用需标定的三氯化钛标准滴定溶液滴定到红色转变为绿色,即为终点。整个滴定过程应在二氧化碳气流保护下操作,同时以45mI水代替重铬酸钾游液以相同的方法做一空白试验。
A3.3三氯化钛标准滴定溶液浓度的表述三氯化钛标准滴定溶液的浓度(c)按式(A1)计算:V,xci
(Al)
式中:V,一一重铬酸钾标准滴定溶液体积,mL;一滴定被重铬酸钾标准滴定溶液氧化成高铁所耗用的三氯化钛标准滴定溶液的体积,mI;Va
滴定空白耗用三氯化钛标推滴定溶液的体积,mL:Va
重铬酸钾标准滴定溶液的实际浓度,mal/L。注:以上标定需在分析样品时即时标定。427
小提示:此标准内容仅展示完整标准里的部分截取内容,若需要完整标准请到上方自行免费下载完整标准文档。